
15/06/2026
Blog
From Benchmark to Product Readiness: Supporting the Latvian Genome Reference on MeluXina
By Xavier Besseron (LuxProvide)
Large-scale genome sequencing projects are transforming medical research and leading the way for more personalised healthcare. However, processing thousands of human genomes requires workflows that can scale efficiently across HPC infrastructures.
As part of the EPICURE support programme, LuxProvide worked with the Latvian Biomedical Research and Study Centre to optimise a whole-genome sequencing workflow on the MeluXina supercomputer. By the end of the collaboration, the team reduced execution time by more than three times and transformed an initially constrained setup into a scalable pipeline ready for large-scale genomic analysis.
The project “Exploring additional computational resources for the Latvian genome reference” contributes to broader European efforts such as the Genome of Europe project.
Starting point: Ambition meets computational constraints
When the project team approached EPICURE, they already had a functional analysis workflow based on the nf-core Sarek pipeline running with Nextflow. While the workflow followed community best practices, execution remained confined to a single compute node, resulting in long runtimes and inefficient resource utilisation.
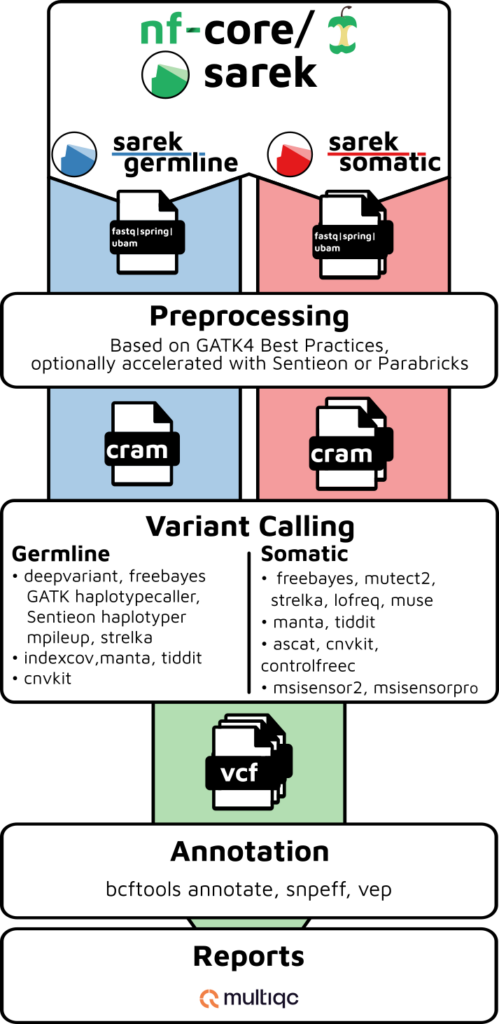
Overview of a generic Sarek pipeline (source)
The challenge was no longer functional correctness, but scalability. The workflow needed to evolve from a working setup into a production-ready solution capable of supporting large-scale genomic analyses.
A step-by-step optimisation strategy
As part of the EPICURE support team at LuxProvide, my role was to help the project explore and optimise MeluXina’s capabilities through iterative benchmarking, workflow optimisation, and performance analysis aligned with the project’s scientific goals.
Rather than applying isolated fixes, the work followed a progressive approach: running benchmarks, analysing results together, validating configuration changes, and testing scalability step by step. By combining performance engineering, workflow expertise, and practical HPC knowledge, while staying closely aligned with the scientific goals, the improvements were not only effective but also reproducible and reusable for future large-scale analyses.
Identifying the main bottlenecks
We began by profiling the workflow on MeluXina’s CPU partition, using Nextflow’s built-in reporting and profiling tools, to understand where time and resources were being spent.
This initial assessment revealed several limiting factors:
- – Key tasks dominate the total execution time.
- – Low CPU utilisation for some long-running steps.
- – High I/O pressure on shared storage.
- – Execution confined to a single compute node due to the default executor configuration.
This diagnostic phase was essential to guide the optimisation process and prioritise the changes with the greatest potential impact.
Scaling beyond a single node
Rather than jumping straight to advanced solutions, we first investigated alternative I/O strategies. Different storage tiers and scratch configurations were evaluated to better understand how the workflow interacted with the HPC environment. Although these changes produced only modest gains, they provided valuable insight into the behaviour of the pipeline under different execution conditions.
Next, we addressed scalability. By moving away from the single-node execution model, we introduced the HyperQueue executor, which enabled efficient multi-node execution within a single allocation. This significantly improved resource utilisation and laid the groundwork for scaling to larger, real-world datasets.
Throughout the process, configurations were carefully documented and validated to ensure long-term reproducibility.
Unlocking GPU Acceleration with Parabricks
The most substantial performance gains came after enabling GPU acceleration with NVIDIA Parabricks for the most computationally intensive alignment tasks.
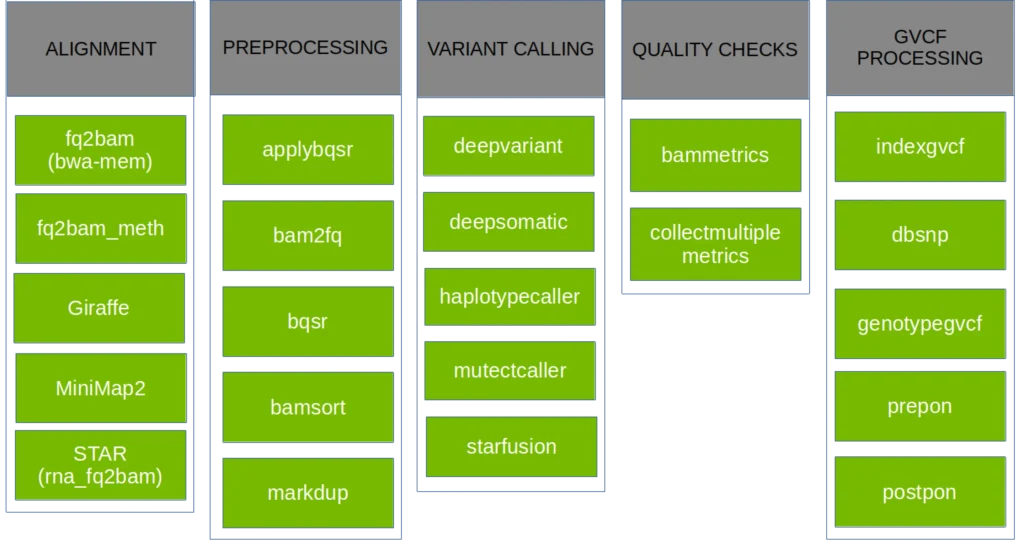
Tools supported by the NVIDIA Parabricks software suite (source)
Integrating GPU acceleration into the workflow required several adjustments, including:
- – Selecting compatible software versions;
- – Fixing missing reference and model files.
- – Debugging workflow errors introduced by changes in pipeline behaviour.
- – Optimising memory usage and execution concurrency on GPU nodes.
Fortunately, support for the Parabricks aligner was introduced in a recent version of the nf-core Sarek pipeline during the final stage of the project, which simplified knowledge transfer and improved usability for the research team.
Lessons learned from the optimisation process
By the end of the project, the optimised workflow achieved speed-ups exceeding a factor of three compared to the initial configuration.
More importantly, the workflow is now able to:
- – Efficiently exploit multi-node execution.
- – Leverage GPU resources where they provide real value.
- – Support large-scale genomic analysis in a stable and reproducible way.
Using GPU resources and NVIDIA Parabricks, the team achieved a speed-up of 3.1× compared to the original CPU-only configuration.
Beyond raw performance improvements, the collaboration also highlighted the importance of early profiling, iterative benchmarking, and close interaction between HPC experts and researchers. Many of the most effective optimisations came not from hardware changes alone, but from understanding how the workflow interacted with the HPC environment.
As a result, the project is now in a much stronger position to scale future analyses and apply for larger EuroHPC access calls with a clearer understanding of its computational requirements.
Looking Ahead
There are still clear opportunities for further optimisation, particularly for downstream analysis steps that dominate runtime once alignment is accelerated. Nevertheless, the project has now moved beyond constrained experimentation and into a phase of scalable production analysis.
The next phase of this project has already started with new objectives: benchmarking large-cohort genomic analyses and tackling hybrid executions that combine CPU and GPU nodes to make optimal use of all available resources.




{kind=link}
{kind=link}
{kind=link}
{kind=link}